| MitImpact id |
MI.7641 |
MI.7642 |
| Chr |
chrM |
chrM |
| Start |
9682 |
9682 |
| Ref |
T |
T |
| Alt |
C |
A |
| Gene symbol |
MT-CO3 |
MT-CO3 |
| Extended annotation |
mitochondrially encoded cytochrome c oxidase III |
mitochondrially encoded cytochrome c oxidase III |
| Gene position |
476 |
476 |
| Gene start |
9207 |
9207 |
| Gene end |
9990 |
9990 |
| Gene strand |
+ |
+ |
| Codon substitution |
ATA/ACA |
ATA/AAA |
| AA position |
159 |
159 |
| AA ref |
M |
M |
| AA alt |
T |
K |
| Functional effect general |
missense |
missense |
| Functional effect detailed |
missense |
missense |
| OMIM id |
516050 |
516050 |
| HGVS |
NC_012920.1:g.9682T>C |
NC_012920.1:g.9682T>A |
| HGNC id |
7422 |
7422 |
| Respiratory Chain complex |
IV |
IV |
| Ensembl gene id |
ENSG00000198938 |
ENSG00000198938 |
| Ensembl transcript id |
ENST00000362079 |
ENST00000362079 |
| Ensembl protein id |
ENSP00000354982 |
ENSP00000354982 |
| Uniprot id |
P00414 |
P00414 |
| Uniprot name |
COX3_HUMAN |
COX3_HUMAN |
| Ncbi gene id |
4514 |
4514 |
| Ncbi protein id |
YP_003024032.1 |
YP_003024032.1 |
| PhyloP 100V |
1.211 |
1.211 |
| PhyloP 470Way |
0.666 |
0.666 |
| PhastCons 100V |
0.002 |
0.002 |
| PhastCons 470Way |
0.26 |
0.26 |
| PolyPhen2 |
benign |
benign |
| PolyPhen2 score |
0.0 |
0.05 |
| SIFT |
neutral |
neutral |
| SIFT score |
0.65 |
0.47 |
| SIFT4G |
Tolerated |
Damaging |
| SIFT4G score |
0.421 |
0.004 |
| VEST |
Neutral |
Pathogenic |
| VEST pvalue |
0.13 |
0.03 |
| VEST FDR |
0.4 |
0.35 |
| Mitoclass.1 |
neutral |
neutral |
| SNPDryad |
Neutral |
Pathogenic |
| SNPDryad score |
0.51 |
0.95 |
| MutationTaster |
. |
. |
| MutationTaster score |
. |
. |
| MutationTaster converted rankscore |
. |
. |
| MutationTaster model |
. |
. |
| MutationTaster AAE |
. |
. |
| fathmm |
. |
. |
| fathmm score |
. |
. |
| fathmm converted rankscore |
. |
. |
| AlphaMissense |
likely_benign |
likely_pathogenic |
| AlphaMissense score |
0.148 |
0.8866 |
| CADD |
Neutral |
Neutral |
| CADD score |
-0.415395 |
2.342562 |
| CADD phred |
0.355 |
18.45 |
| PROVEAN |
Tolerated |
Tolerated |
| PROVEAN score |
1.17 |
-0.88 |
| MutationAssessor |
neutral |
medium |
| MutationAssessor score |
-0.885 |
2.845 |
| EFIN SP |
Neutral |
Damaging |
| EFIN SP score |
0.784 |
0.584 |
| EFIN HD |
Neutral |
Neutral |
| EFIN HD score |
0.798 |
0.412 |
| MLC |
Neutral |
Neutral |
| MLC score |
0.28351138 |
0.28351138 |
| PANTHER score |
. |
. |
| PhD-SNP score |
. |
. |
| APOGEE1 |
Neutral |
Neutral |
| APOGEE1 score |
0.23 |
0.34 |
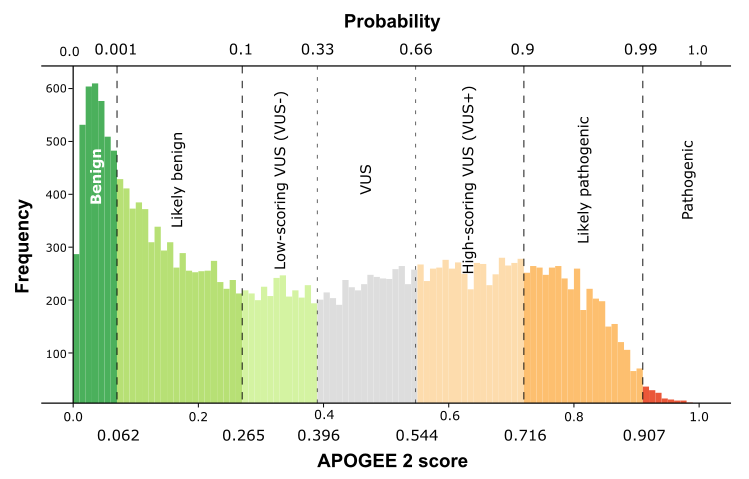
| APOGEE2 |
Benign |
Likely-benign |
| APOGEE2 score |
0.0315833974242117 |
0.218759461182717 |
| CAROL |
neutral |
neutral |
| CAROL score |
0.34 |
0.48 |
| Condel |
deleterious |
deleterious |
| Condel score |
0.83 |
0.71 |
| COVEC WMV |
neutral |
neutral |
| COVEC WMV score |
-6 |
-6 |
| MtoolBox |
neutral |
neutral |
| MtoolBox DS |
0.14 |
0.26 |
| DEOGEN2 |
. |
. |
| DEOGEN2 score |
. |
. |
| DEOGEN2 converted rankscore |
. |
. |
| Meta-SNP |
. |
. |
| Meta-SNP score |
. |
. |
| PolyPhen2 transf |
high impact |
medium impact |
| PolyPhen2 transf score |
2.05 |
0.37 |
| SIFT_transf |
medium impact |
medium impact |
| SIFT transf score |
0.35 |
0.16 |
| MutationAssessor transf |
low impact |
medium impact |
| MutationAssessor transf score |
-2.03 |
-0.06 |
| CHASM |
Neutral |
Neutral |
| CHASM pvalue |
0.17 |
0.21 |
| CHASM FDR |
0.8 |
0.8 |
| ClinVar id |
693205.0 |
. |
| ClinVar Allele id |
681741.0 |
. |
| ClinVar CLNDISDB |
MONDO:MONDO:0009723,MedGen:C0023264,OMIM:256000,Orphanet:506 |
. |
| ClinVar CLNDN |
Leigh_syndrome |
. |
| ClinVar CLNSIG |
Benign |
. |
| MITOMAP Disease Clinical info |
. |
. |
| MITOMAP Disease Status |
. |
. |
| MITOMAP Disease Hom/Het |
./. |
./. |
| MITOMAP General GenBank Freq |
0.139% |
. |
| MITOMAP General GenBank Seqs |
85 |
. |
| MITOMAP General Curated refs |
21978175;24467713;11938495;12509511;12840039;15791543;16172508 |
. |
| MITOMAP Variant Class |
polymorphism |
. |
| gnomAD 3.1 AN |
56425.0 |
. |
| gnomAD 3.1 AC Homo |
791.0 |
. |
| gnomAD 3.1 AF Hom |
0.0140186 |
. |
| gnomAD 3.1 AC Het |
5.0 |
. |
| gnomAD 3.1 AF Het |
8.86132e-05 |
. |
| gnomAD 3.1 filter |
PASS |
. |
| HelixMTdb AC Hom |
614.0 |
. |
| HelixMTdb AF Hom |
0.003132925 |
. |
| HelixMTdb AC Het |
6.0 |
. |
| HelixMTdb AF Het |
3.06149e-05 |
. |
| HelixMTdb mean ARF |
0.63829 |
. |
| HelixMTdb max ARF |
0.77512 |
. |
| ToMMo 54KJPN AC |
9 |
. |
| ToMMo 54KJPN AF |
0.000166 |
. |
| ToMMo 54KJPN AN |
54302 |
. |
| COSMIC 90 |
. |
. |
| dbSNP 156 id |
. |
. |