This case-study shows how to assess the functionality of a variant using MitImpact. It will pass through all sections of a typical result page.
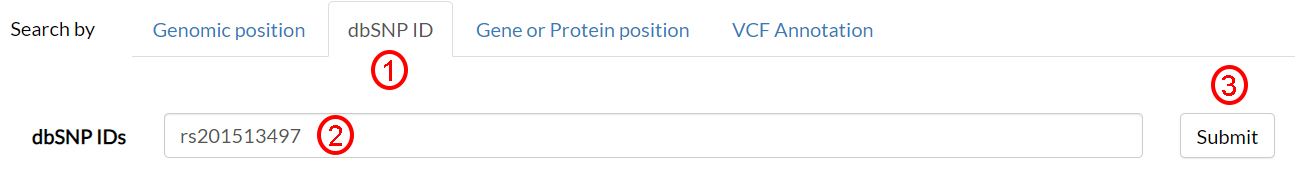
Starting from a dbSNP ID of interest (e.g. rs201513497), any associated information can be sought by selecting dbSNP ID (1), filling the text field accordingly (2), and clicking on Submit (3).
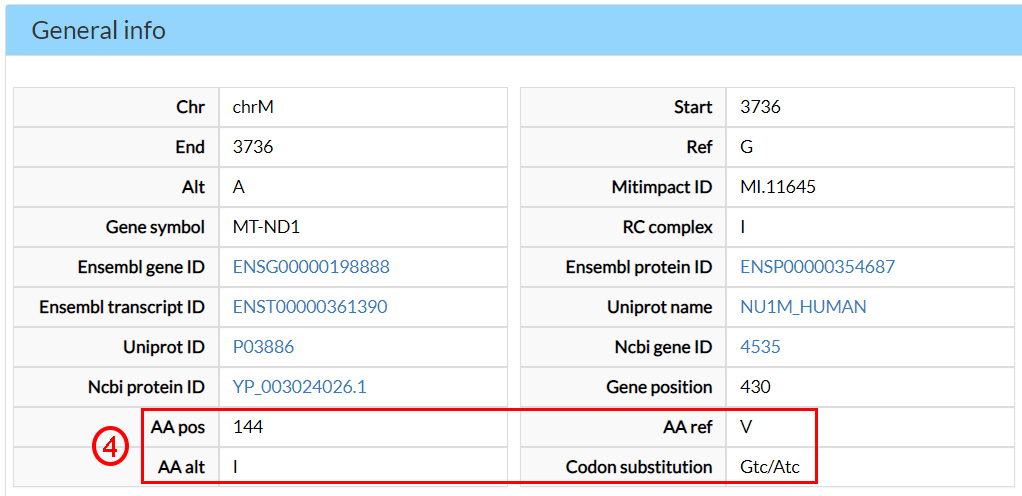
In the next page, results are split into different sections. In the former, the General info section, you can find several annotations, including the alternate amino acid (4).
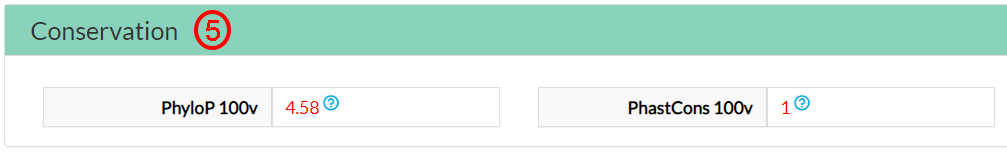
The Conservation scores indicate that this amino acid is evolutionary conserved (5).
Two predictors on 16 (6) and two meta-predictors on 7 (7) agree that this variant amino acid (144 Isoleucine) could be damaging.
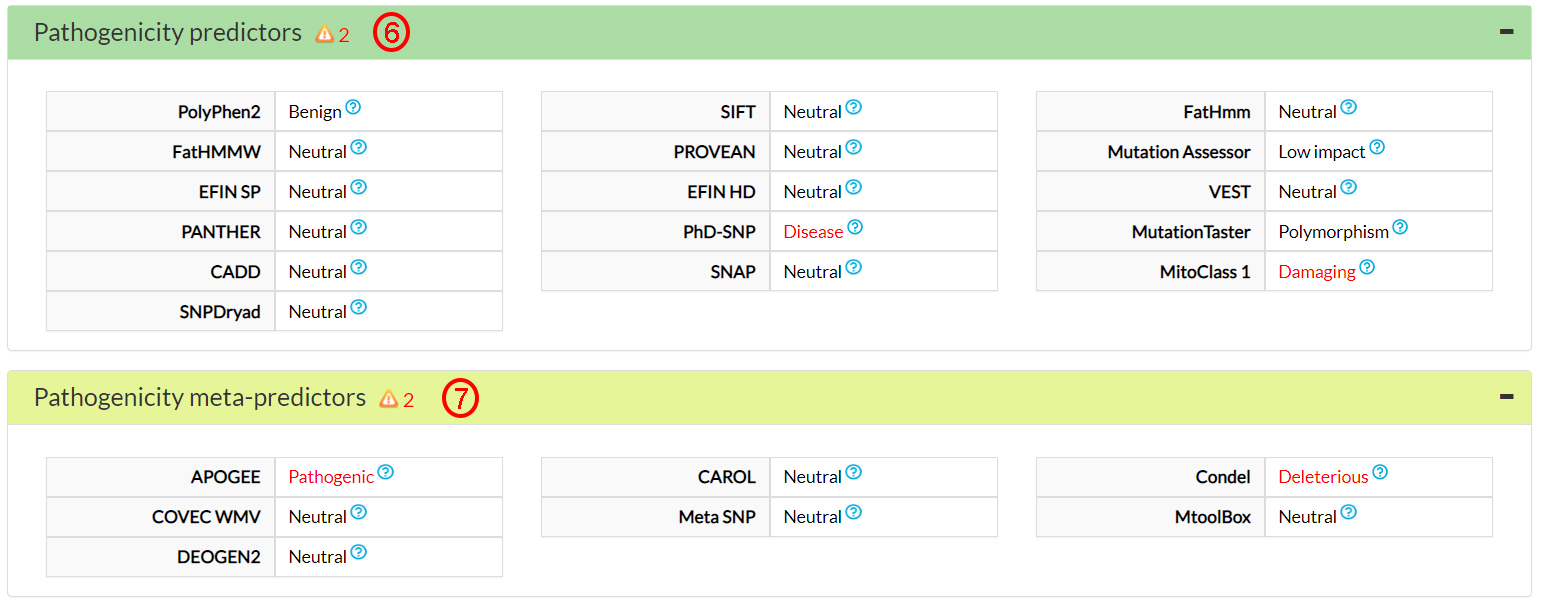
This is confirmed by 3 cancer-specific predictors (8).
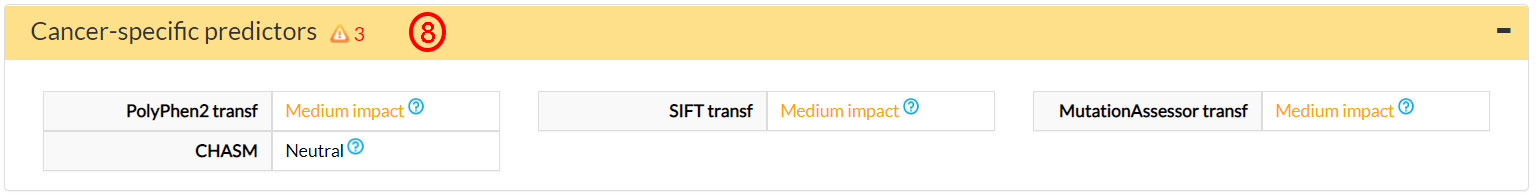
In the Databases of Phenotypes, you can find information about the association between the V144I variant and the clinical phenotypes retrieved from Clinvar and MITOMAP databases. This variant is associated with LHON and Leigh syndromes (9).
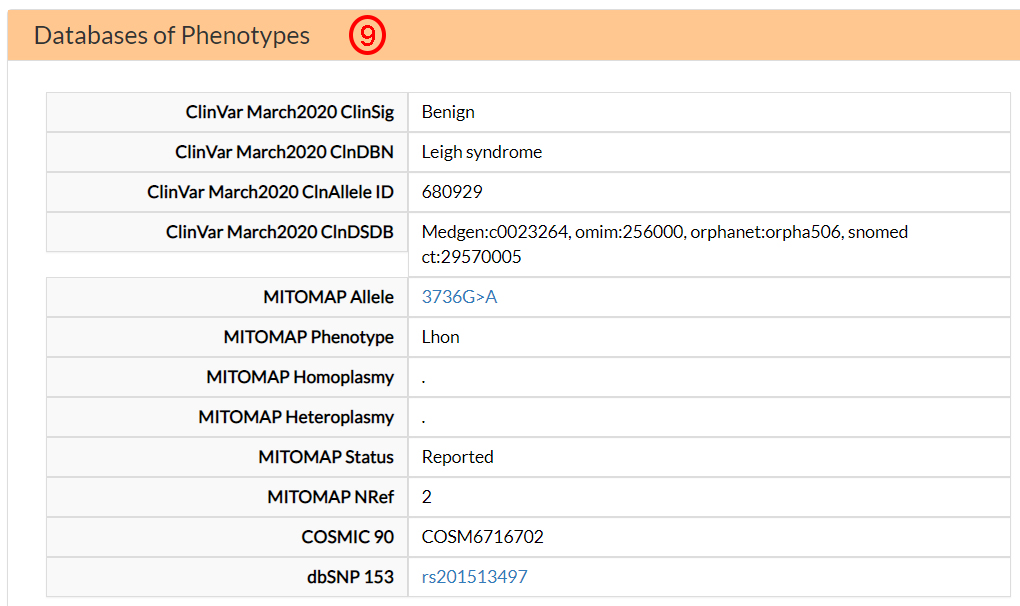
In the Residue interaction section, you can find
information on epistasis related mechanisms, specifically based on co-variation (EVmutation,
Site A-B InterP and Site A-B IntraP) and energetic parameters (ΔΔG intra, ΔΔG Intra interface
and ΔΔG inter).
From the EVmutation option menu, you can select one of six predicted intra-protein (ND1) co-variants sites
(10). Moreover, from the Site A-B InterP option
menu, you can find three possible covariant amino acids in two different mitochondrial proteins (ND3 and ND6)
(11). Contrary to EVmutation, Site A-B IntraP does not report
any pair of sites that are estimated to co-vary based on this prediction system.
It is important to remark that co-variation scores
(i.e., EV score, mfDCA, and corrected MI) are not meant to prioritize one pair over another,
since all reported pairs of co-varying variants were selected from the top scoring pairs obtained as explained in the
Description page.
The last three menu options in the previous figure report energetic variations (ΔΔG) of variants hitting
amino acids at co-varying sites which are located inter or intra-protein. In this particular case,
the variant 144V in ND1 was found to interact with the amino acid N10 in ND3
(12). The energetic changes caused by the mutants in
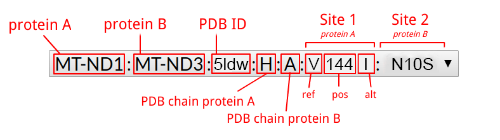
these positions were assessed and reported in the option menu. Selecting, for example, the 10S variant in ND3
(PDB ID: 5ldw), one obtains information on the energetic variation caused by each single variants (ΔΔG 1st and ΔΔG 2nd)
and of both variants (ΔΔG both, i.e., V144I and N10S)
(13).
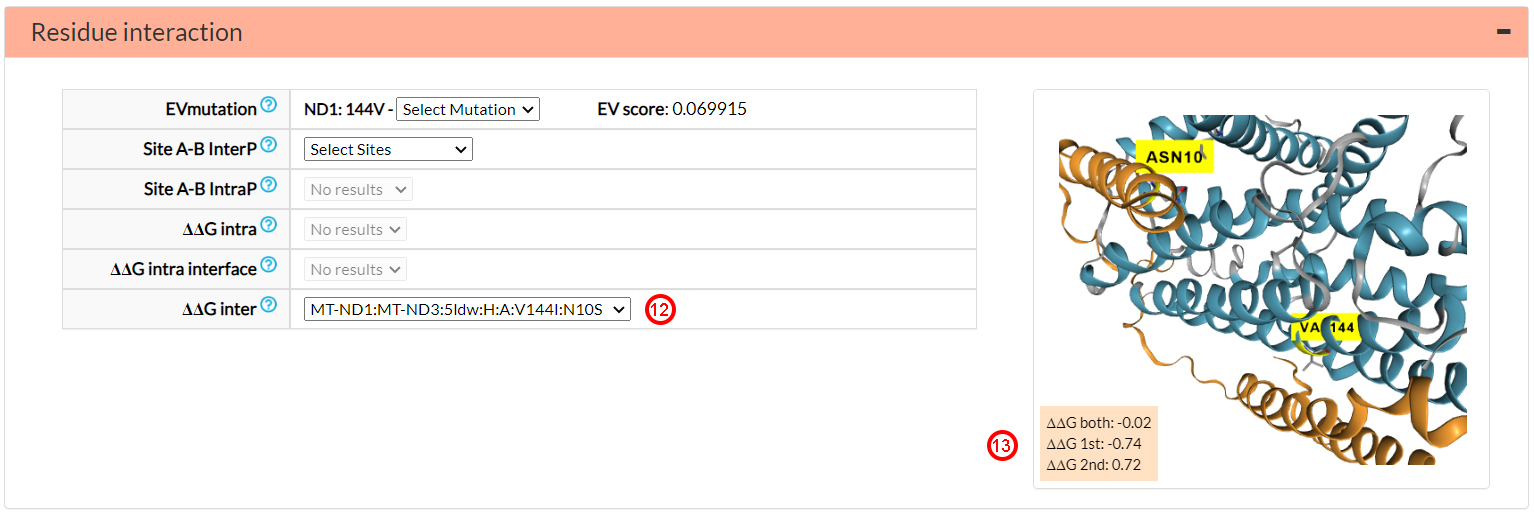
In general, the more extreme, positively or negatively, a ΔΔG value,
the more energetically destabilizing or stabilizing a variant can be considered. A ΔΔG both value greater than that of the single
mutants (-0.74 and 0.72) can be associated to a possibly harmful interaction between the two
variants. On the contrary as in this case, a ΔΔG value of the double mutant close to zero (-0.02) can
be associated to an energetic compensatory interaction.
A handy legend of the selected option menu item follows.
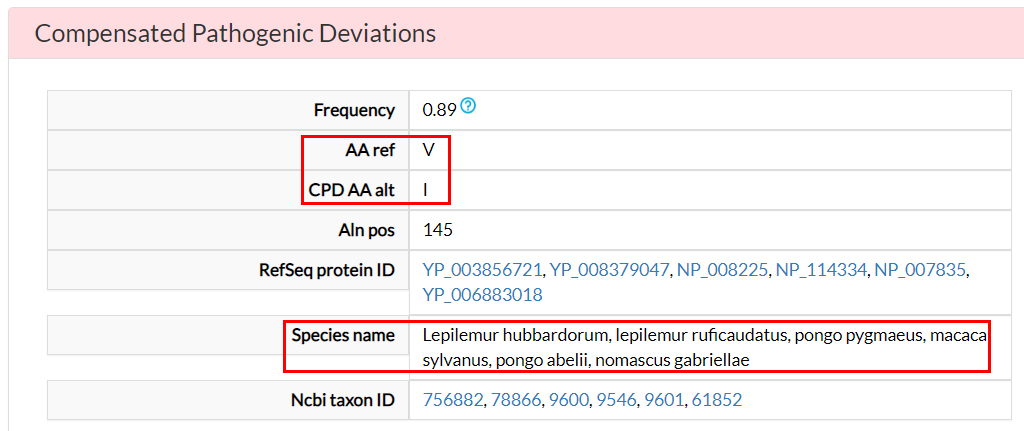
In the Compensated Pathogenic Deviations section,
the amino acid substitutions that are reported as pathogenic in the human population, but occur as wild-type
residues in non-human ortholog proteins, are reported. These are tagged as potentially compensated
variants.
For the V144I variant, there is a potential compensating
variant (18) in at least six species.
The impact of the variant V144I on the ND1-ND3 interaction, as well as any putative compensatory effects,
can also be investigated in the
Molecular Dynamics section (19) of the website.

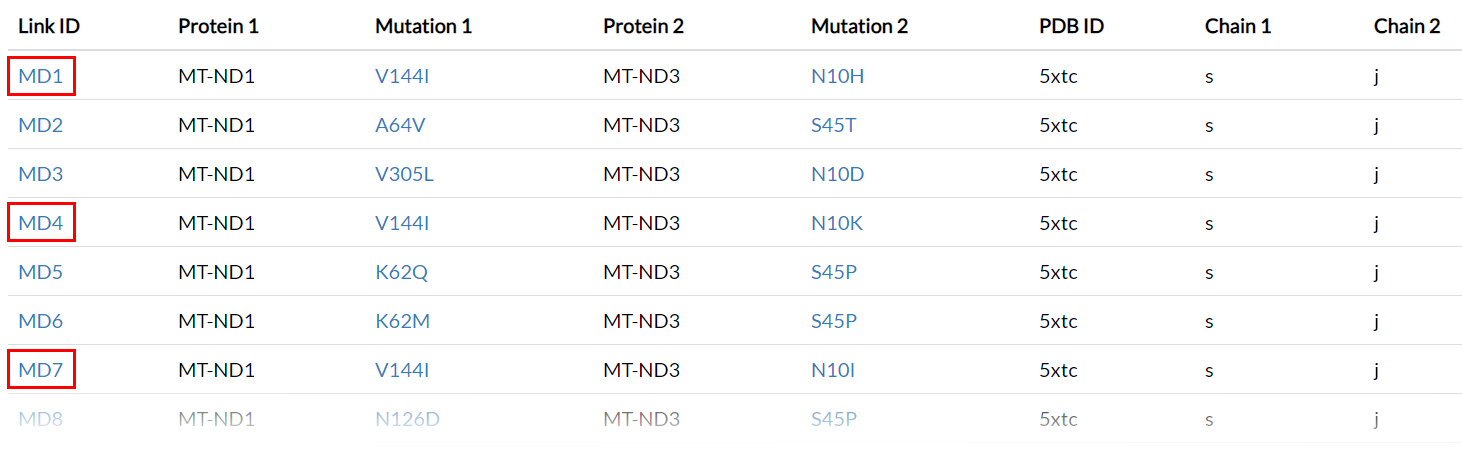
The webpages with IDs: MD1, MD4, MD7, MD17, MD23, MD25 and MD38
collect results obtained by simulation of the amino acid variants pairs involving V144I in ND1 in combination
with multiple possible variants affecting the site 10 in ND3.
These Link IDs could also be reached by clicking on the Dynamics links button
(20) on top-right of the V144I result page.
Clicking on the MT-DN3_5xtc_s_j_VA144I_NB10S (21) link,
we can examine the molecular dynamics behaviour of the V144I-N10S variant pairs found as possibly compensatory
in the previous Residue interaction section.
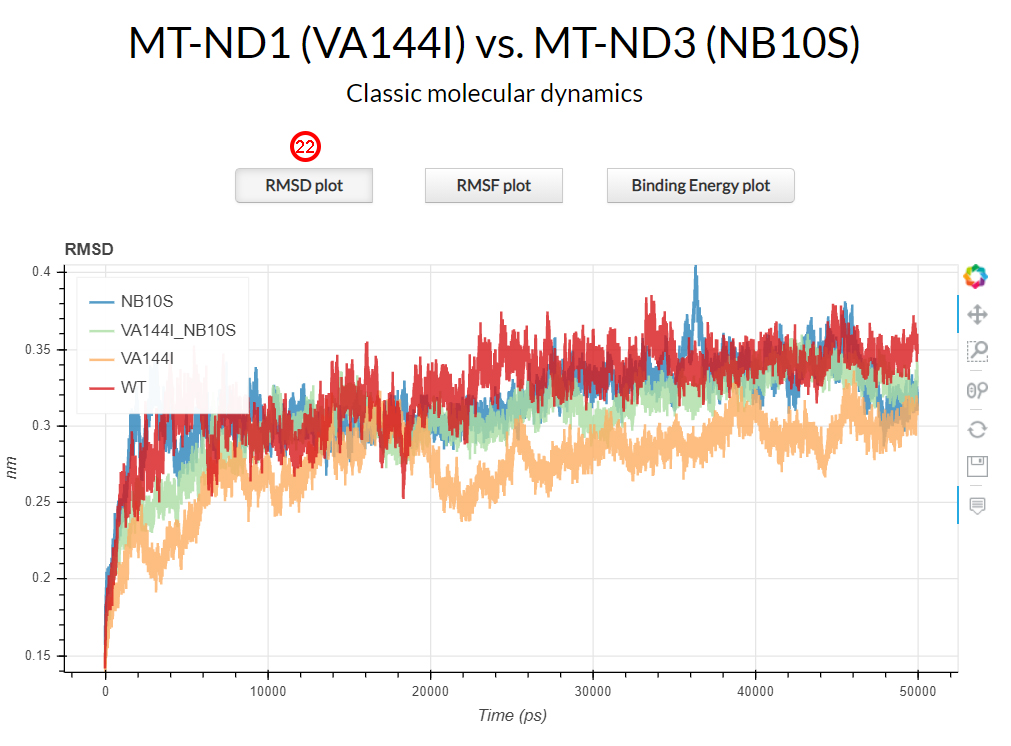
Root-mean-square deviation (RMSD) measures the average distance between all heavy atoms (in this case Cα
atomic coordinates) with respect to the X-ray structure. The RMSD of all heavy atoms is shown in the first
section of results page by clicking the corresponding RMSD plot button (22).
The RMSD plot shows how the V144I mutant (in orange) exhibited lower RMSD values during the simulation time
with respect to the wild-type protein complex (in red). N10S (in blue) and the double mutant V144I-N10S (in green)
showed comparable RMSD values with the wild-type.
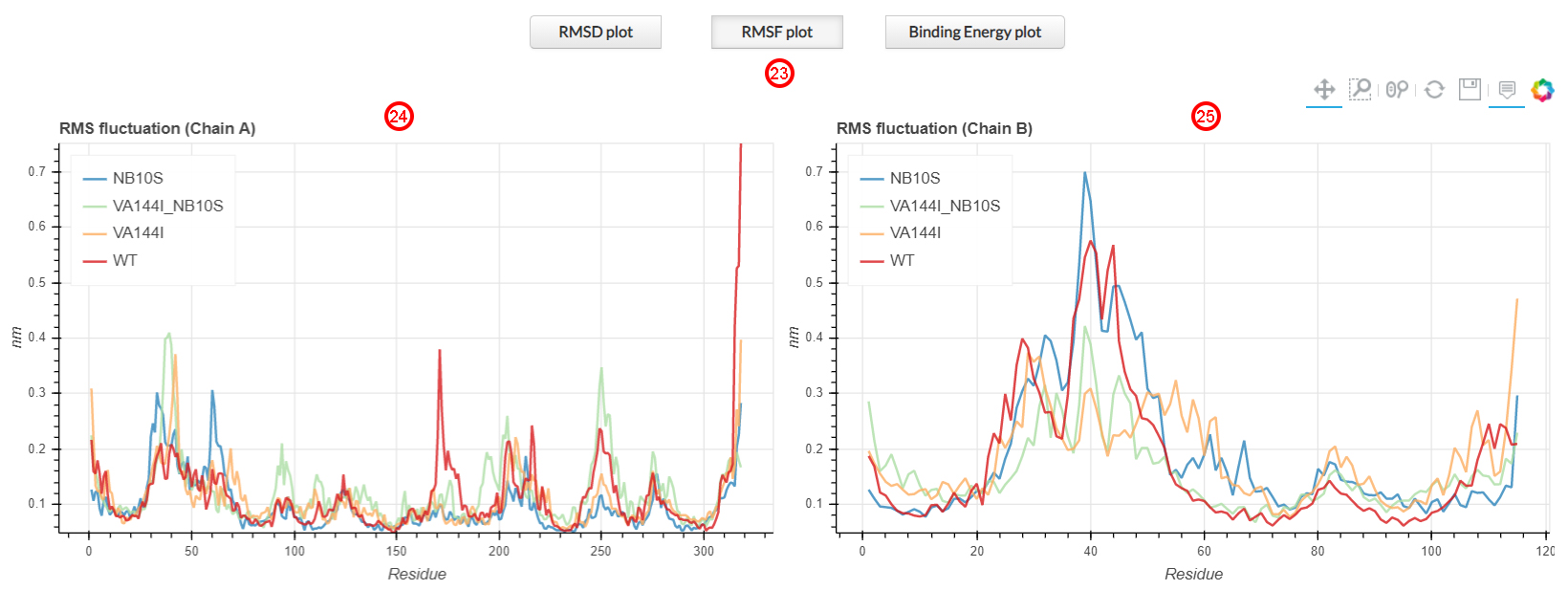
Root mean square fluctuation (RMSF) measures the deviation over time of the positions of the Cα atomic
coordinates of each residue with respect to the X-ray structure. The RMSF of all heavy atoms is shown in the
first section of the results page by clicking the corresponding RMSF plot button (23).
On the left (24), we can observe the ND1 (chain A) residues fluctuations,
while on the right (25) we observe the fluctuations of the ND3 residues (chain B).
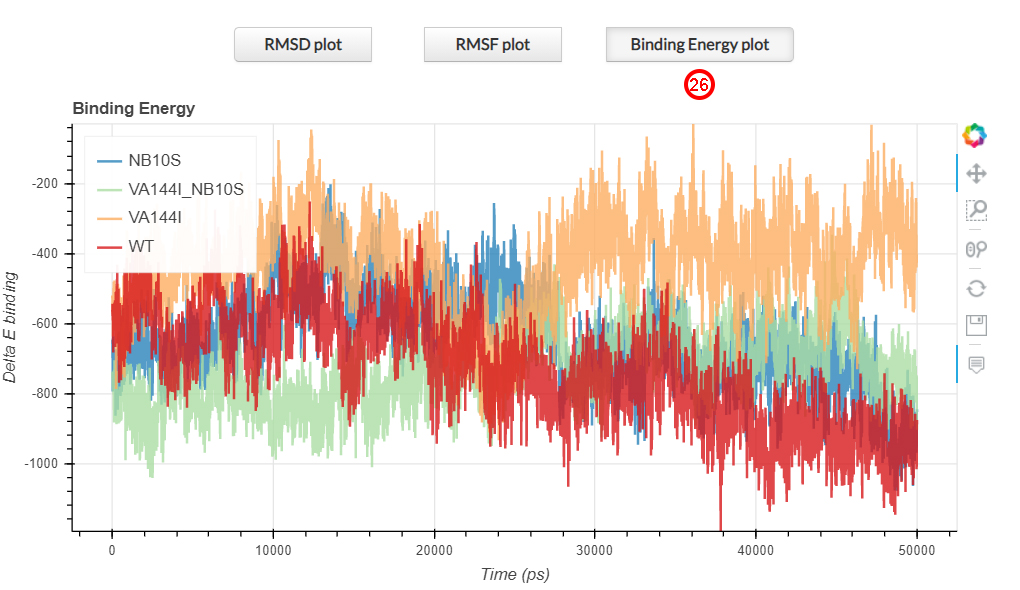
The binding energy plot represent the interaction energy between two proteins. The lowere the measured
binding energy, the higher the interaction between two protein. This plot is shown in the first section of
the results page by clicking on the corresponding Binding Energy plot button (26).
The V144I mutation (in orange) causes an alteration of the binding energy values during the entire simulation time,
compared with the wild-type, while the simultaneous presence of both variants on the ND1-ND3 interface
(green line), as well as the N10S variant (blue line), resembles the wild-type condition (red line).
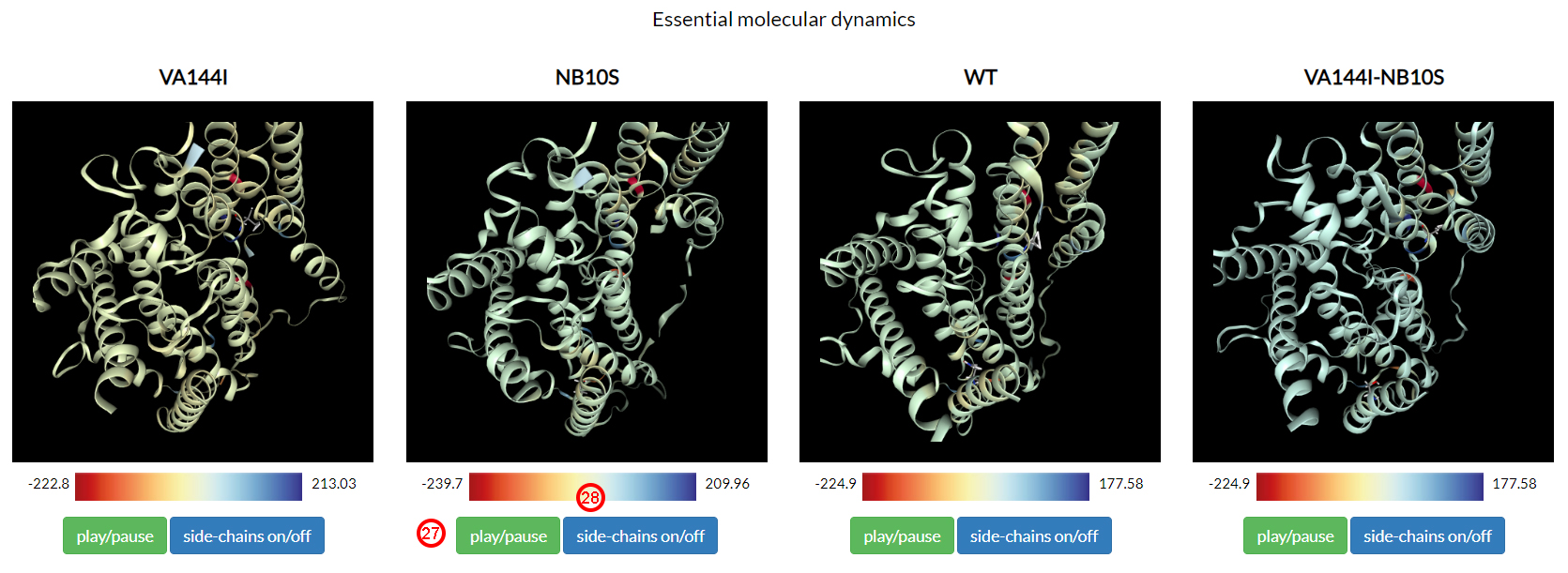
Essential dynamical motions from all the above systems are shown in the second section of the results page.
In brief, the essential molecular dynamics analysis allows to separate the large collective protein motions
associated with functional properties from the global slow and uninteresting motion.
Clicking on each play/pause button (27), we can observe how the essential motions
of VA144I recall a separation movement, consistent with the decreasing of the binding energy observed above.
On the contrary, wild-type and double mutant showed a comparable overall dynamics.
Interface residues are colored according to their binding energy contribution during the entire simulation time.
Colors range from red to blue (28), which correspond to the minimum and
maximum per-complex ΔΔG values of the residues at the interaction interface.
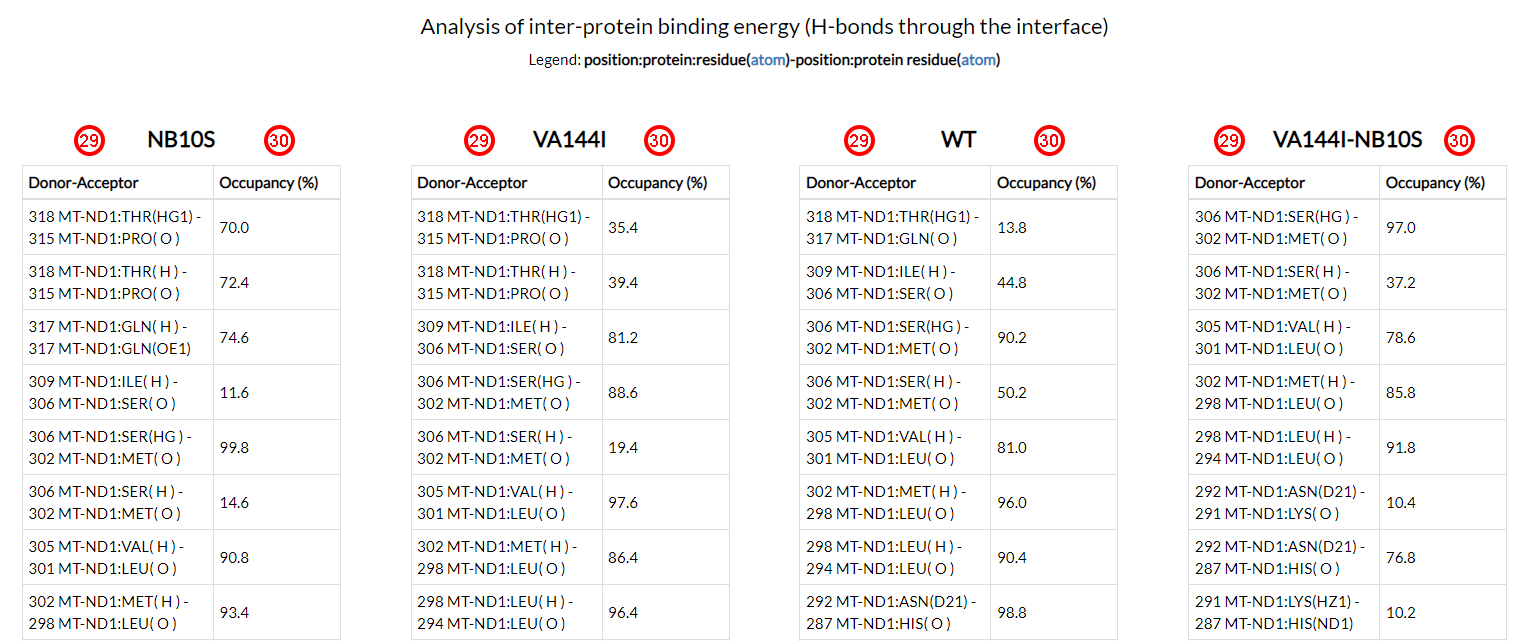
In the last section of the results page, all hydrogen bonds (acceptor and donor atoms of each residue
position (29)) and the corresponding resident times (as a percentage
of the total simulation time (30))
were reported for each of the four simulations.
These results do not highlight any key-player hydrogen bond. Howecer, an overall redistribution of the
h-bonds network, is evident when analyzing the resident times of the residues located at the
interface between the two interacting proteins.
The selected variant rs201513497, 3736G>A, causes an amino acid change in an evolutionary conserved residue,
but a non concordant deleterious effect was provided by pathogenicity predictors.
Inter/intra-protein co-variation analyses showed how this site co-varies with ND3 (in position 10)
and ND6 (in positions 109 and 116).
This result underlies V144I as an important site of the ND1 protein that may play a role in the
complex organization of the OXPHOS system. The energetic and structural analyses of the co-varying sites
demonstrate how this specific substitution is predicted to be stabilized by a co-occurring residue located
at position 10 of the interacting ND3 protein. The occurrence of a compensating pathogenic variant
for V144I suggests that interacting amino acids could modulate the molecular impact of this amino acid variant.
Finally, the analysis of the MD simulations of the mutant ND1 V144I combined with the N10S variant on the
interacting ND3 protein showed how the co-occurence of both mutations could partially restore
the fundamental dynamical properties, such as stability and binding affinity, that recall
the dynamical behaviours of the wild-type protein system.