| ~ | 9175 (C/A) | 9175 (C/G) |
|---|---|---|
| ~ | 9175 (CTA/ATA) | 9175 (CTA/GTA) |
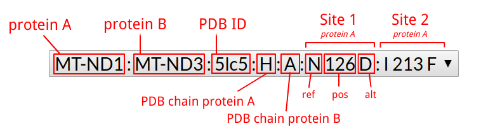
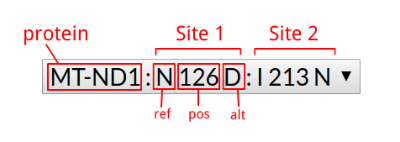
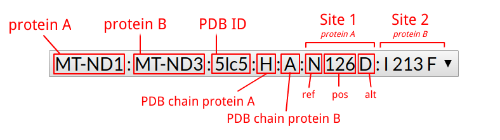
| MitImpact id | MI.1382 | MI.1381 |
| Chr | chrM | chrM |
| Start | 9175 | 9175 |
| Ref | C | C |
| Alt | A | G |
| Gene symbol | MT-ATP6 | MT-ATP6 |
| Extended annotation | mitochondrially encoded ATP synthase membrane subunit 6 | mitochondrially encoded ATP synthase membrane subunit 6 |
| Gene position | 649 | 649 |
| Gene start | 8527 | 8527 |
| Gene end | 9207 | 9207 |
| Gene strand | + | + |
| Codon substitution | CTA/ATA | CTA/GTA |
| AA position | 217 | 217 |
| AA ref | L | L |
| AA alt | M | V |
| Functional effect general | missense | missense |
| Functional effect detailed | missense | missense |
| OMIM id | 516060 | 516060 |
| HGVS | NC_012920.1:g.9175C>A | NC_012920.1:g.9175C>G |
| HGNC id | 7414 | 7414 |
| Respiratory Chain complex | V | V |
| Ensembl gene id | ENSG00000198899 | ENSG00000198899 |
| Ensembl transcript id | ENST00000361899 | ENST00000361899 |
| Ensembl protein id | ENSP00000354632 | ENSP00000354632 |
| Uniprot id | P00846 | P00846 |
| Uniprot name | ATP6_HUMAN | ATP6_HUMAN |
| Ncbi gene id | 4508 | 4508 |
| Ncbi protein id | YP_003024031.1 | YP_003024031.1 |
| PhyloP 100V | -0.192 | -0.192 |
| PhyloP 470Way | 0.724 | 0.724 |
| PhastCons 100V | 0 | 0 |
| PhastCons 470Way | 0.795 | 0.795 |
| PolyPhen2 | probably_damaging | probably_damaging |
| PolyPhen2 score | 1.0 | 0.99 |
| SIFT | deleterious | deleterious |
| SIFT score | 0 | 0.01 |
| SIFT4G | Damaging | Damaging |
| SIFT4G score | 0.0 | 0.0 |
| VEST | Neutral | Neutral |
| VEST pvalue | 0.16 | 0.2 |
| VEST FDR | 0.65 | 0.65 |
| Mitoclass.1 | damaging | damaging |
| SNPDryad | Pathogenic | Neutral |
| SNPDryad score | 0.96 | 0.87 |
| MutationTaster | . | Polymorphism |
| MutationTaster score | . | 0.818404 |
| MutationTaster converted rankscore | . | 0.28913 |
| MutationTaster model | . | simple_aae |
| MutationTaster AAE | . | L217V |
| fathmm | . | Tolerated |
| fathmm score | . | 2.32 |
| fathmm converted rankscore | . | 0.16640 |
| AlphaMissense | likely_pathogenic | likely_pathogenic |
| AlphaMissense score | 0.7785 | 0.8663 |
| CADD | Deleterious | Deleterious |
| CADD score | 3.856233 | 3.52406 |
| CADD phred | 23.5 | 23.1 |
| PROVEAN | Tolerated | Damaging |
| PROVEAN score | -1.78 | -2.67 |
| MutationAssessor | . | high |
| MutationAssessor score | . | 4.985 |
| EFIN SP | Damaging | Damaging |
| EFIN SP score | 0.432 | 0.356 |
| EFIN HD | Neutral | Neutral |
| EFIN HD score | 0.596 | 0.514 |
| MLC | Neutral | Neutral |
| MLC score | 0.08775424 | 0.08775424 |
| PANTHER score | . | . |
| PhD-SNP score | . | . |
| APOGEE1 | Pathogenic | Pathogenic |
| APOGEE1 score | 0.62 | 0.61 |
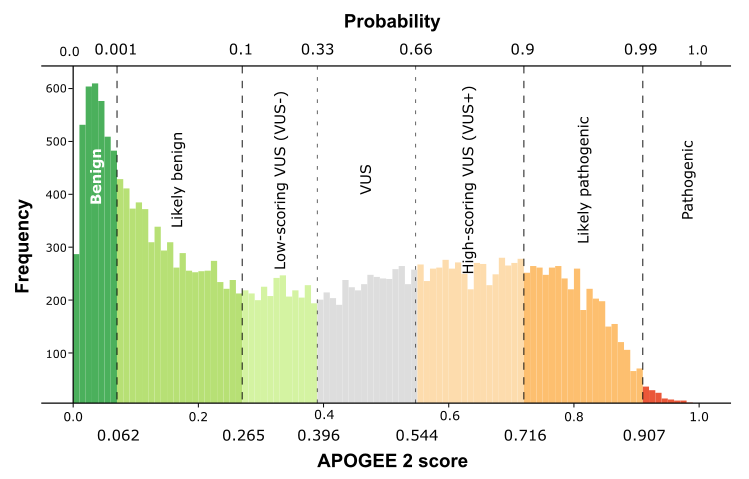
| APOGEE2 | VUS- | VUS+ |
| APOGEE2 score | 0.366250668285401 | 0.551760265175148 |
| CAROL | deleterious | deleterious |
| CAROL score | 1 | 1 |
| Condel | neutral | neutral |
| Condel score | 0 | 0.01 |
| COVEC WMV | deleterious | deleterious |
| COVEC WMV score | 6 | 6 |
| MtoolBox | deleterious | deleterious |
| MtoolBox DS | 0.82 | 0.85 |
| DEOGEN2 | . | Tolerated |
| DEOGEN2 score | . | 0.417708 |
| DEOGEN2 converted rankscore | . | 0.77027 |
| Meta-SNP | . | . |
| Meta-SNP score | . | . |
| PolyPhen2 transf | low impact | low impact |
| PolyPhen2 transf score | -3.6 | -2.65 |
| SIFT_transf | low impact | medium impact |
| SIFT transf score | -1.4 | -0.84 |
| MutationAssessor transf | high impact | high impact |
| MutationAssessor transf score | 2.82 | 2.34 |
| CHASM | Neutral | Neutral |
| CHASM pvalue | 0.58 | 0.68 |
| CHASM FDR | 0.9 | 0.9 |
| ClinVar id | . | . |
| ClinVar Allele id | . | . |
| ClinVar CLNDISDB | . | . |
| ClinVar CLNDN | . | . |
| ClinVar CLNSIG | . | . |
| MITOMAP Disease Clinical info | . | . |
| MITOMAP Disease Status | . | . |
| MITOMAP Disease Hom/Het | ./. | ./. |
| MITOMAP General GenBank Freq | 0.0049% | . |
| MITOMAP General GenBank Seqs | 3 | . |
| MITOMAP General Curated refs | 17072496 | . |
| MITOMAP Variant Class | polymorphism | . |
| gnomAD 3.1 AN | 56434.0 | . |
| gnomAD 3.1 AC Homo | 1.0 | . |
| gnomAD 3.1 AF Hom | 1.77198e-05 | . |
| gnomAD 3.1 AC Het | 0.0 | . |
| gnomAD 3.1 AF Het | 0.0 | . |
| gnomAD 3.1 filter | PASS | . |
| HelixMTdb AC Hom | 12.0 | . |
| HelixMTdb AF Hom | 6.12298e-05 | . |
| HelixMTdb AC Het | 1.0 | . |
| HelixMTdb AF Het | 5.1024836e-06 | . |
| HelixMTdb mean ARF | 0.67665 | . |
| HelixMTdb max ARF | 0.67665 | . |
| ToMMo 54KJPN AC | . | . |
| ToMMo 54KJPN AF | . | . |
| ToMMo 54KJPN AN | . | . |
| COSMIC 90 | . | . |
| dbSNP 156 id | . | . |