| MitImpact id |
MI.689 |
MI.690 |
| Chr |
chrM |
chrM |
| Start |
8851 |
8851 |
| Ref |
T |
T |
| Alt |
C |
G |
| Gene symbol |
MT-ATP6 |
MT-ATP6 |
| Extended annotation |
mitochondrially encoded ATP synthase membrane subunit 6 |
mitochondrially encoded ATP synthase membrane subunit 6 |
| Gene position |
325 |
325 |
| Gene start |
8527 |
8527 |
| Gene end |
9207 |
9207 |
| Gene strand |
+ |
+ |
| Codon substitution |
TGA/CGA |
TGA/GGA |
| AA position |
109 |
109 |
| AA ref |
W |
W |
| AA alt |
R |
G |
| Functional effect general |
missense |
missense |
| Functional effect detailed |
missense |
missense |
| OMIM id |
516060 |
516060 |
| HGVS |
NC_012920.1:g.8851T>C |
NC_012920.1:g.8851T>G |
| HGNC id |
7414 |
7414 |
| Respiratory Chain complex |
V |
V |
| Ensembl gene id |
ENSG00000198899 |
ENSG00000198899 |
| Ensembl transcript id |
ENST00000361899 |
ENST00000361899 |
| Ensembl protein id |
ENSP00000354632 |
ENSP00000354632 |
| Uniprot id |
P00846 |
P00846 |
| Uniprot name |
ATP6_HUMAN |
ATP6_HUMAN |
| Ncbi gene id |
4508 |
4508 |
| Ncbi protein id |
YP_003024031.1 |
YP_003024031.1 |
| PhyloP 100V |
5.983 |
5.983 |
| PhyloP 470Way |
0.666 |
0.666 |
| PhastCons 100V |
1 |
1 |
| PhastCons 470Way |
0.736 |
0.736 |
| PolyPhen2 |
probably_damaging |
probably_damaging |
| PolyPhen2 score |
1.0 |
0.99 |
| SIFT |
deleterious |
deleterious |
| SIFT score |
0 |
0 |
| SIFT4G |
Damaging |
Damaging |
| SIFT4G score |
0.0 |
0.0 |
| VEST |
Neutral |
Neutral |
| VEST pvalue |
0.28 |
0.24 |
| VEST FDR |
0.65 |
0.65 |
| Mitoclass.1 |
damaging |
damaging |
| SNPDryad |
Pathogenic |
Pathogenic |
| SNPDryad score |
0.97 |
1 |
| MutationTaster |
. |
. |
| MutationTaster score |
. |
. |
| MutationTaster converted rankscore |
. |
. |
| MutationTaster model |
. |
. |
| MutationTaster AAE |
. |
. |
| fathmm |
. |
. |
| fathmm score |
. |
. |
| fathmm converted rankscore |
. |
. |
| AlphaMissense |
likely_pathogenic |
likely_pathogenic |
| AlphaMissense score |
0.987 |
0.8008 |
| CADD |
Deleterious |
Deleterious |
| CADD score |
3.556674 |
3.870712 |
| CADD phred |
23.1 |
23.5 |
| PROVEAN |
Damaging |
Damaging |
| PROVEAN score |
-12.92 |
-11.98 |
| MutationAssessor |
. |
high |
| MutationAssessor score |
. |
5.32 |
| EFIN SP |
Damaging |
Neutral |
| EFIN SP score |
0.414 |
0.618 |
| EFIN HD |
Damaging |
Neutral |
| EFIN HD score |
0.254 |
0.416 |
| MLC |
Neutral |
Neutral |
| MLC score |
0.04828294 |
0.04828294 |
| PANTHER score |
. |
. |
| PhD-SNP score |
. |
. |
| APOGEE1 |
Pathogenic |
Neutral |
| APOGEE1 score |
0.69 |
0.5 |
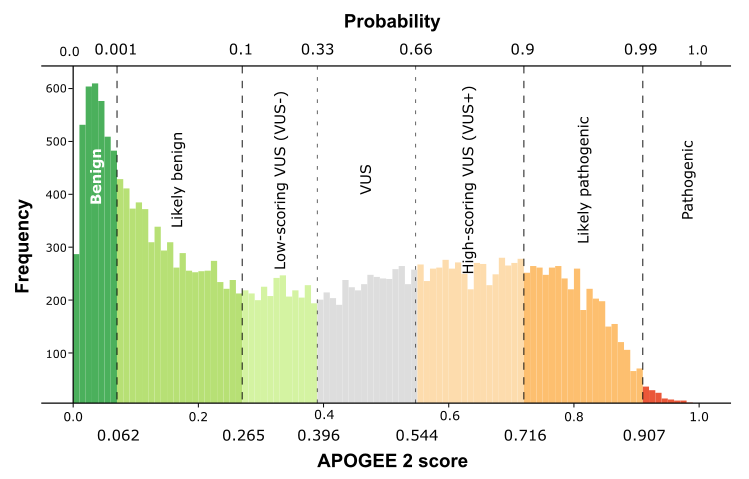
| APOGEE2 |
Likely-pathogenic |
Likely-pathogenic |
| APOGEE2 score |
0.867435890954745 |
0.72895471332079 |
| CAROL |
deleterious |
deleterious |
| CAROL score |
1 |
1 |
| Condel |
neutral |
neutral |
| Condel score |
0 |
0.01 |
| COVEC WMV |
deleterious |
deleterious |
| COVEC WMV score |
6 |
6 |
| MtoolBox |
deleterious |
deleterious |
| MtoolBox DS |
0.9 |
0.84 |
| DEOGEN2 |
. |
. |
| DEOGEN2 score |
. |
. |
| DEOGEN2 converted rankscore |
. |
. |
| Meta-SNP |
. |
. |
| Meta-SNP score |
. |
. |
| PolyPhen2 transf |
low impact |
low impact |
| PolyPhen2 transf score |
-3.6 |
-2.65 |
| SIFT_transf |
low impact |
low impact |
| SIFT transf score |
-1.4 |
-1.4 |
| MutationAssessor transf |
high impact |
high impact |
| MutationAssessor transf score |
2.72 |
2.72 |
| CHASM |
Neutral |
Neutral |
| CHASM pvalue |
0.24 |
0.28 |
| CHASM FDR |
0.9 |
0.9 |
| ClinVar id |
9645.0 |
. |
| ClinVar Allele id |
24684.0 |
. |
| ClinVar CLNDISDB |
MONDO:MONDO:0044970,MedGen:C0751651,Orphanet:68380|MONDO:MONDO:0010774,MedGen:C1839022,OMIM:500003|MedGen:CN517202|Human_Phenotype_Ontology:HP:0001086,Human_Phenotype_Ontology:HP:0001112,MONDO:MONDO:0010788,MedGen:C0917796,OMIM:535000,Orphanet:104|MONDO:MONDO:0009723,MedGen:C0023264,OMIM:256000,Orphanet:506 |
. |
| ClinVar CLNDN |
Mitochondrial_disease|Striatonigral_degeneration,_infantile,_mitochondrial|not_provided|Leber_optic_atrophy|Leigh_syndrome |
. |
| ClinVar CLNSIG |
Uncertain_significance |
. |
| MITOMAP Disease Clinical info |
BSN / Leigh syndrome |
. |
| MITOMAP Disease Status |
Cfrm [VUS*] |
. |
| MITOMAP Disease Hom/Het |
+/+ |
./. |
| MITOMAP General GenBank Freq |
0.0065% |
. |
| MITOMAP General GenBank Seqs |
4 |
. |
| MITOMAP General Curated refs |
18620007;23206802;24002810;21470976;30763462;21457906;8554662;29253894;32652755 |
. |
| MITOMAP Variant Class |
disease |
. |
| gnomAD 3.1 AN |
56428.0 |
56433.0 |
| gnomAD 3.1 AC Homo |
2.0 |
0.0 |
| gnomAD 3.1 AF Hom |
3.54434e-05 |
0.0 |
| gnomAD 3.1 AC Het |
0.0 |
0.0 |
| gnomAD 3.1 AF Het |
0.0 |
0.0 |
| gnomAD 3.1 filter |
PASS |
npg |
| HelixMTdb AC Hom |
10.0 |
. |
| HelixMTdb AF Hom |
5.1024836e-05 |
. |
| HelixMTdb AC Het |
4.0 |
. |
| HelixMTdb AF Het |
2.0409934e-05 |
. |
| HelixMTdb mean ARF |
0.29899 |
. |
| HelixMTdb max ARF |
0.59701 |
. |
| ToMMo 54KJPN AC |
1 |
. |
| ToMMo 54KJPN AF |
1.8e-05 |
. |
| ToMMo 54KJPN AN |
54302 |
. |
| COSMIC 90 |
. |
. |
| dbSNP 156 id |
. |
. |