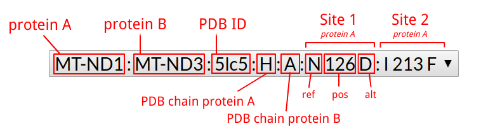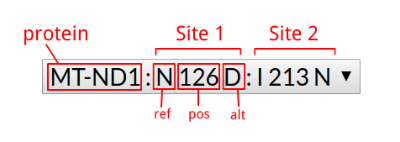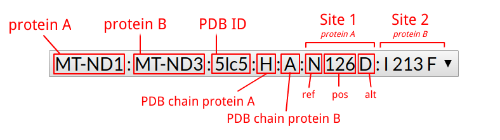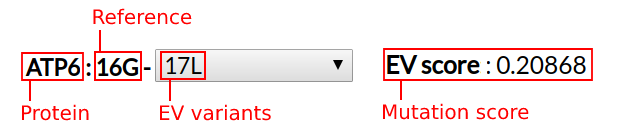
| ~ | 4768 (T/C) | 4768 (T/A) |
|---|---|---|
| ~ | 4768 (ATA/ACA) | 4768 (ATA/AAA) |
| MitImpact id | MI.13392 | MI.13393 |
| Chr | chrM | chrM |
| Start | 4768 | 4768 |
| Ref | T | T |
| Alt | C | A |
| Gene symbol | MT-ND2 | MT-ND2 |
| Extended annotation | mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 2 | mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 2 |
| Gene position | 299 | 299 |
| Gene start | 4470 | 4470 |
| Gene end | 5511 | 5511 |
| Gene strand | + | + |
| Codon substitution | ATA/ACA | ATA/AAA |
| AA position | 100 | 100 |
| AA ref | M | M |
| AA alt | T | K |
| Functional effect general | missense | missense |
| Functional effect detailed | missense | missense |
| OMIM id | 516001 | 516001 |
| HGVS | NC_012920.1:g.4768T>C | NC_012920.1:g.4768T>A |
| HGNC id | 7456 | 7456 |
| Respiratory Chain complex | I | I |
| Ensembl gene id | ENSG00000198763 | ENSG00000198763 |
| Ensembl transcript id | ENST00000361453 | ENST00000361453 |
| Ensembl protein id | ENSP00000355046 | ENSP00000355046 |
| Uniprot id | P03891 | P03891 |
| Uniprot name | NU2M_HUMAN | NU2M_HUMAN |
| Ncbi gene id | 4536 | 4536 |
| Ncbi protein id | YP_003024027.1 | YP_003024027.1 |
| PhyloP 100V | -0.4 | -0.4 |
| PhyloP 470Way | 0.458 | 0.458 |
| PhastCons 100V | 0 | 0 |
| PhastCons 470Way | 0.007 | 0.007 |
| PolyPhen2 | benign | benign |
| PolyPhen2 score | 0.0 | 0.04 |
| SIFT | neutral | neutral |
| SIFT score | 0.36 | 0.23 |
| SIFT4G | Tolerated | Damaging |
| SIFT4G score | 0.235 | 0.002 |
| VEST | Neutral | Pathogenic |
| VEST pvalue | 0.09 | 0.03 |
| VEST FDR | 0.35 | 0.35 |
| Mitoclass.1 | neutral | neutral |
| SNPDryad | Neutral | Neutral |
| SNPDryad score | 0.03 | 0.62 |
| MutationTaster | . | . |
| MutationTaster score | . | . |
| MutationTaster converted rankscore | . | . |
| MutationTaster model | . | . |
| MutationTaster AAE | . | . |
| fathmm | . | . |
| fathmm score | . | . |
| fathmm converted rankscore | . | . |
| AlphaMissense | likely_benign | likely_pathogenic |
| AlphaMissense score | 0.1115 | 0.9676 |
| CADD | Neutral | Neutral |
| CADD score | -0.464901 | 2.206545 |
| CADD phred | 0.268 | 17.55 |
| PROVEAN | Tolerated | Damaging |
| PROVEAN score | -2.01 | -3.68 |
| MutationAssessor | neutral | medium |
| MutationAssessor score | 0.395 | 3.235 |
| EFIN SP | Neutral | Neutral |
| EFIN SP score | 0.944 | 0.87 |
| EFIN HD | Neutral | Neutral |
| EFIN HD score | 0.992 | 0.376 |
| MLC | Neutral | Neutral |
| MLC score | 0.2262961 | 0.2262961 |
| PANTHER score | . | . |
| PhD-SNP score | . | . |
| APOGEE1 | Neutral | Neutral |
| APOGEE1 score | 0.31 | 0.35 |
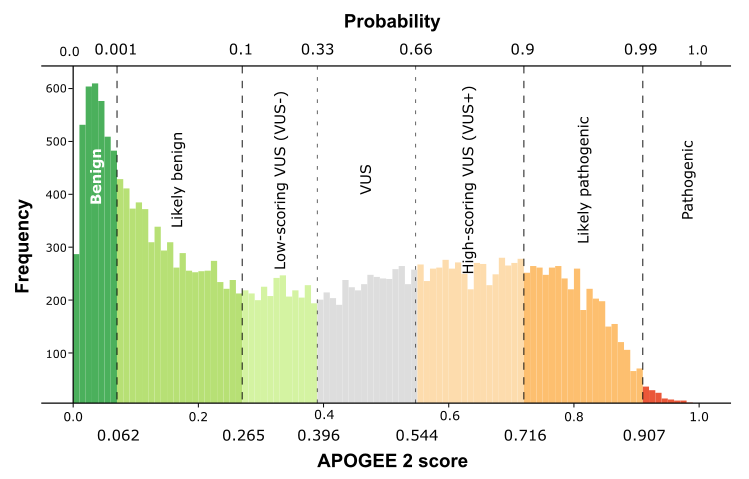
| APOGEE2 | Likely-benign | VUS |
| APOGEE2 score | 0.0718448013915129 | 0.483778676410232 |
| CAROL | neutral | neutral |
| CAROL score | 0.64 | 0.75 |
| Condel | deleterious | deleterious |
| Condel score | 0.68 | 0.6 |
| COVEC WMV | neutral | neutral |
| COVEC WMV score | -6 | -3 |
| MtoolBox | neutral | neutral |
| MtoolBox DS | 0.16 | 0.28 |
| DEOGEN2 | . | . |
| DEOGEN2 score | . | . |
| DEOGEN2 converted rankscore | . | . |
| Meta-SNP | . | . |
| Meta-SNP score | . | . |
| PolyPhen2 transf | medium impact | medium impact |
| PolyPhen2 transf score | 1.95 | 0.47 |
| SIFT_transf | medium impact | medium impact |
| SIFT transf score | 0.07 | -0.08 |
| MutationAssessor transf | medium impact | medium impact |
| MutationAssessor transf score | -0.59 | 1.15 |
| CHASM | Neutral | Neutral |
| CHASM pvalue | 0.19 | 0.3 |
| CHASM FDR | 0.8 | 0.8 |
| ClinVar id | 235413.0 | . |
| ClinVar Allele id | 237098.0 | . |
| ClinVar CLNDISDB | MedGen:CN517202 | . |
| ClinVar CLNDN | not_provided | . |
| ClinVar CLNSIG | Uncertain_significance | . |
| MITOMAP Disease Clinical info | . | . |
| MITOMAP Disease Status | . | . |
| MITOMAP Disease Hom/Het | ./. | ./. |
| MITOMAP General GenBank Freq | 0.0131% | . |
| MITOMAP General GenBank Seqs | 8 | . |
| MITOMAP General Curated refs | . | . |
| MITOMAP Variant Class | polymorphism | . |
| gnomAD 3.1 AN | 56431.0 | . |
| gnomAD 3.1 AC Homo | 8.0 | . |
| gnomAD 3.1 AF Hom | 0.000141766 | . |
| gnomAD 3.1 AC Het | 1.0 | . |
| gnomAD 3.1 AF Het | 1.77208e-05 | . |
| gnomAD 3.1 filter | PASS | . |
| HelixMTdb AC Hom | 27.0 | . |
| HelixMTdb AF Hom | 0.00013776706 | . |
| HelixMTdb AC Het | 5.0 | . |
| HelixMTdb AF Het | 2.5512418e-05 | . |
| HelixMTdb mean ARF | 0.3525 | . |
| HelixMTdb max ARF | 0.60494 | . |
| ToMMo 54KJPN AC | 6 | . |
| ToMMo 54KJPN AF | 0.00011 | . |
| ToMMo 54KJPN AN | 54302 | . |
| COSMIC 90 | . | . |
| dbSNP 156 id | . | . |