| MitImpact id |
MI.24032 |
MI.24033 |
MI.24031 |
| Chr |
chrM |
chrM |
chrM |
| Start |
14598 |
14598 |
14598 |
| Ref |
T |
T |
T |
| Alt |
C |
A |
G |
| Gene symbol |
MT-ND6 |
MT-ND6 |
MT-ND6 |
| Extended annotation |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 6 |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 6 |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 6 |
| Gene position |
76 |
76 |
76 |
| Gene start |
14149 |
14149 |
14149 |
| Gene end |
14673 |
14673 |
14673 |
| Gene strand |
- |
- |
- |
| Codon substitution |
ATT/GTT |
ATT/TTT |
ATT/CTT |
| AA position |
26 |
26 |
26 |
| AA ref |
I |
I |
I |
| AA alt |
V |
F |
L |
| Functional effect general |
missense |
missense |
missense |
| Functional effect detailed |
missense |
missense |
missense |
| OMIM id |
516006 |
516006 |
516006 |
| HGVS |
NC_012920.1:g.14598T>C |
NC_012920.1:g.14598T>A |
NC_012920.1:g.14598T>G |
| HGNC id |
7462 |
7462 |
7462 |
| Respiratory Chain complex |
I |
I |
I |
| Ensembl gene id |
ENSG00000198695 |
ENSG00000198695 |
ENSG00000198695 |
| Ensembl transcript id |
ENST00000361681 |
ENST00000361681 |
ENST00000361681 |
| Ensembl protein id |
ENSP00000354665 |
ENSP00000354665 |
ENSP00000354665 |
| Uniprot id |
P03923 |
P03923 |
P03923 |
| Uniprot name |
NU6M_HUMAN |
NU6M_HUMAN |
NU6M_HUMAN |
| Ncbi gene id |
4541 |
4541 |
4541 |
| Ncbi protein id |
YP_003024037.1 |
YP_003024037.1 |
YP_003024037.1 |
| PhyloP 100V |
0.263 |
0.263 |
0.263 |
| PhyloP 470Way |
0.666 |
0.666 |
0.666 |
| PhastCons 100V |
0.009 |
0.009 |
0.009 |
| PhastCons 470Way |
0.997 |
0.997 |
0.997 |
| PolyPhen2 |
probably_damaging |
probably_damaging |
probably_damaging |
| PolyPhen2 score |
1 |
1 |
1 |
| SIFT |
neutral |
neutral |
neutral |
| SIFT score |
0.45 |
0.59 |
0.61 |
| SIFT4G |
Tolerated |
Damaging |
Damaging |
| SIFT4G score |
0.387 |
0.041 |
0.023 |
| VEST |
Neutral |
Neutral |
Neutral |
| VEST pvalue |
0.58 |
0.51 |
0.39 |
| VEST FDR |
0.65 |
0.6 |
0.5 |
| Mitoclass.1 |
damaging |
damaging |
damaging |
| SNPDryad |
Neutral |
Pathogenic |
Neutral |
| SNPDryad score |
0.15 |
0.93 |
0.62 |
| MutationTaster |
Disease |
. |
Disease |
| MutationTaster score |
1 |
. |
1 |
| MutationTaster converted rankscore |
0.81001 |
. |
0.81001 |
| MutationTaster model |
without_aae |
. |
without_aae |
| MutationTaster AAE |
. |
. |
. |
| fathmm |
Tolerated |
Tolerated |
Tolerated |
| fathmm score |
2.37 |
2.1 |
2.21 |
| fathmm converted rankscore |
0.15964 |
0.19990 |
0.18414 |
| AlphaMissense |
likely_benign |
likely_pathogenic |
likely_benign |
| AlphaMissense score |
0.0915 |
0.5657 |
0.2824 |
| CADD |
Neutral |
Deleterious |
Neutral |
| CADD score |
-0.451492 |
4.159179 |
2.351432 |
| CADD phred |
0.29 |
23.8 |
18.5 |
| PROVEAN |
Tolerated |
Damaging |
Tolerated |
| PROVEAN score |
-0.62 |
-3.97 |
-1.99 |
| MutationAssessor |
low |
low |
low |
| MutationAssessor score |
0.985 |
0.92 |
1.88 |
| EFIN SP |
Neutral |
Neutral |
Damaging |
| EFIN SP score |
0.786 |
0.624 |
0.406 |
| EFIN HD |
Neutral |
Neutral |
Neutral |
| EFIN HD score |
0.844 |
0.666 |
0.368 |
| MLC |
Deleterious |
Deleterious |
Deleterious |
| MLC score |
0.75427002 |
0.75427002 |
0.75427002 |
| PANTHER score |
. |
. |
. |
| PhD-SNP score |
. |
. |
. |
| APOGEE1 |
Neutral |
Neutral |
Pathogenic |
| APOGEE1 score |
0.31 |
0.31 |
0.65 |
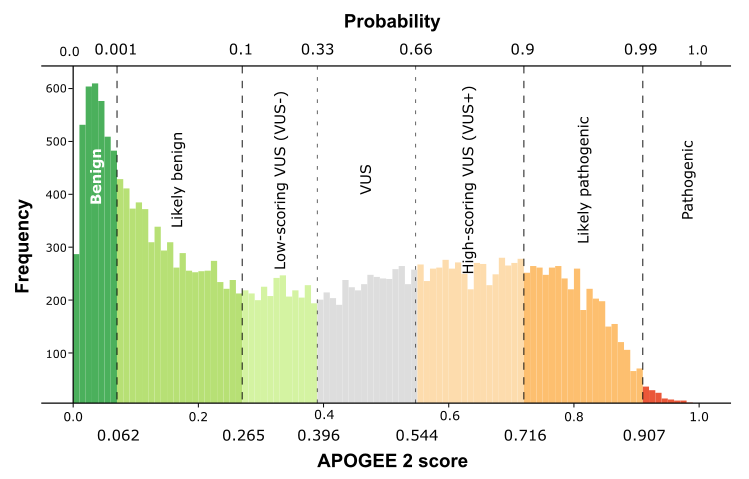
| APOGEE2 |
Benign |
VUS- |
VUS+ |
| APOGEE2 score |
0.0498621622144087 |
0.302875209549539 |
0.629375553785344 |
| CAROL |
deleterious |
deleterious |
deleterious |
| CAROL score |
0.99 |
1.0 |
0.99 |
| Condel |
neutral |
neutral |
neutral |
| Condel score |
0.23 |
0.3 |
0.31 |
| COVEC WMV |
neutral |
deleterious |
deleterious |
| COVEC WMV score |
-2 |
1 |
1 |
| MtoolBox |
deleterious |
deleterious |
deleterious |
| MtoolBox DS |
0.68 |
0.7 |
0.7 |
| DEOGEN2 |
Tolerated |
Damaging |
Tolerated |
| DEOGEN2 score |
0.218413 |
0.531547 |
0.410017 |
| DEOGEN2 converted rankscore |
0.58113 |
0.83798 |
0.76488 |
| Meta-SNP |
. |
. |
. |
| Meta-SNP score |
. |
. |
. |
| PolyPhen2 transf |
low impact |
low impact |
low impact |
| PolyPhen2 transf score |
-3.55 |
-3.55 |
-3.55 |
| SIFT_transf |
medium impact |
medium impact |
medium impact |
| SIFT transf score |
0.16 |
0.29 |
0.31 |
| MutationAssessor transf |
medium impact |
medium impact |
medium impact |
| MutationAssessor transf score |
0.29 |
0.49 |
1.46 |
| CHASM |
Neutral |
Neutral |
Neutral |
| CHASM pvalue |
0.63 |
0.78 |
0.68 |
| CHASM FDR |
0.8 |
0.85 |
0.85 |
| ClinVar id |
374080.0 |
. |
. |
| ClinVar Allele id |
361108.0 |
. |
. |
| ClinVar CLNDISDB |
MONDO:MONDO:0009723,MedGen:C0023264,OMIM:256000,Orphanet:506|Human_Phenotype_Ontology:HP:0001300,MedGen:C0242422|Human_Phenotype_Ontology:HP:0000618,Human_Phenotype_Ontology:HP:0007839,MedGen:C0456909 |
. |
. |
| ClinVar CLNDN |
Leigh_syndrome|Parkinsonism|Blindness |
. |
. |
| ClinVar CLNSIG |
Conflicting_interpretations_of_pathogenicity |
. |
. |
| MITOMAP Disease Clinical info |
PD / LS |
. |
. |
| MITOMAP Disease Status |
Reported [VUS] |
. |
. |
| MITOMAP Disease Hom/Het |
+/- |
./. |
./. |
| MITOMAP General GenBank Freq |
0.0115% |
. |
. |
| MITOMAP General GenBank Seqs |
7 |
. |
. |
| MITOMAP General Curated refs |
24002810;23463613 |
. |
. |
| MITOMAP Variant Class |
polymorphism;disease |
. |
. |
| gnomAD 3.1 AN |
56425.0 |
. |
. |
| gnomAD 3.1 AC Homo |
9.0 |
. |
. |
| gnomAD 3.1 AF Hom |
0.000159504 |
. |
. |
| gnomAD 3.1 AC Het |
3.0 |
. |
. |
| gnomAD 3.1 AF Het |
5.31679e-05 |
. |
. |
| gnomAD 3.1 filter |
PASS |
. |
. |
| HelixMTdb AC Hom |
26.0 |
. |
. |
| HelixMTdb AF Hom |
0.00013266457 |
. |
. |
| HelixMTdb AC Het |
3.0 |
. |
. |
| HelixMTdb AF Het |
1.530745e-05 |
. |
. |
| HelixMTdb mean ARF |
0.19312 |
. |
. |
| HelixMTdb max ARF |
0.30476 |
. |
. |
| ToMMo 54KJPN AC |
11 |
. |
. |
| ToMMo 54KJPN AF |
0.000203 |
. |
. |
| ToMMo 54KJPN AN |
54301 |
. |
. |
| COSMIC 90 |
COSM6716806 |
. |
. |
| dbSNP 156 id |
rs1057518882 |
. |
. |