| MitImpact id |
MI.23813 |
MI.23812 |
| Chr |
chrM |
chrM |
| Start |
14495 |
14495 |
| Ref |
A |
A |
| Alt |
G |
C |
| Gene symbol |
MT-ND6 |
MT-ND6 |
| Extended annotation |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 6 |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 6 |
| Gene position |
179 |
179 |
| Gene start |
14149 |
14149 |
| Gene end |
14673 |
14673 |
| Gene strand |
- |
- |
| Codon substitution |
TTA/TCA |
TTA/TGA |
| AA position |
60 |
60 |
| AA ref |
L |
L |
| AA alt |
S |
W |
| Functional effect general |
missense |
missense |
| Functional effect detailed |
missense |
missense |
| OMIM id |
516006 |
516006 |
| HGVS |
NC_012920.1:g.14495A>G |
NC_012920.1:g.14495A>C |
| HGNC id |
7462 |
7462 |
| Respiratory Chain complex |
I |
I |
| Ensembl gene id |
ENSG00000198695 |
ENSG00000198695 |
| Ensembl transcript id |
ENST00000361681 |
ENST00000361681 |
| Ensembl protein id |
ENSP00000354665 |
ENSP00000354665 |
| Uniprot id |
P03923 |
P03923 |
| Uniprot name |
NU6M_HUMAN |
NU6M_HUMAN |
| Ncbi gene id |
4541 |
4541 |
| Ncbi protein id |
YP_003024037.1 |
YP_003024037.1 |
| PhyloP 100V |
6.872 |
6.872 |
| PhyloP 470Way |
0.819 |
0.819 |
| PhastCons 100V |
1 |
1 |
| PhastCons 470Way |
0.994 |
0.994 |
| PolyPhen2 |
probably_damaging |
probably_damaging |
| PolyPhen2 score |
1 |
1 |
| SIFT |
neutral |
neutral |
| SIFT score |
0.29 |
0.12 |
| SIFT4G |
Damaging |
Damaging |
| SIFT4G score |
0.0 |
0.0 |
| VEST |
Neutral |
Neutral |
| VEST pvalue |
0.19 |
0.15 |
| VEST FDR |
0.45 |
0.45 |
| Mitoclass.1 |
damaging |
damaging |
| SNPDryad |
Pathogenic |
Pathogenic |
| SNPDryad score |
0.97 |
0.98 |
| MutationTaster |
Disease |
. |
| MutationTaster score |
1 |
. |
| MutationTaster converted rankscore |
0.81001 |
. |
| MutationTaster model |
without_aae |
. |
| MutationTaster AAE |
. |
. |
| fathmm |
Tolerated |
. |
| fathmm score |
2.17 |
. |
| fathmm converted rankscore |
0.19020 |
. |
| AlphaMissense |
likely_pathogenic |
likely_pathogenic |
| AlphaMissense score |
0.9348 |
0.9219 |
| CADD |
Deleterious |
Deleterious |
| CADD score |
3.774913 |
3.292697 |
| CADD phred |
23.4 |
22.8 |
| PROVEAN |
Damaging |
Damaging |
| PROVEAN score |
-5.99 |
-5.99 |
| MutationAssessor |
high |
. |
| MutationAssessor score |
3.52 |
. |
| EFIN SP |
Damaging |
Damaging |
| EFIN SP score |
0.232 |
0.502 |
| EFIN HD |
Damaging |
Damaging |
| EFIN HD score |
0.166 |
0.252 |
| MLC |
Neutral |
Neutral |
| MLC score |
0.40364536 |
0.40364536 |
| PANTHER score |
0.815 |
. |
| PhD-SNP score |
0.813 |
. |
| APOGEE1 |
Pathogenic |
Neutral |
| APOGEE1 score |
0.87 |
0.42 |
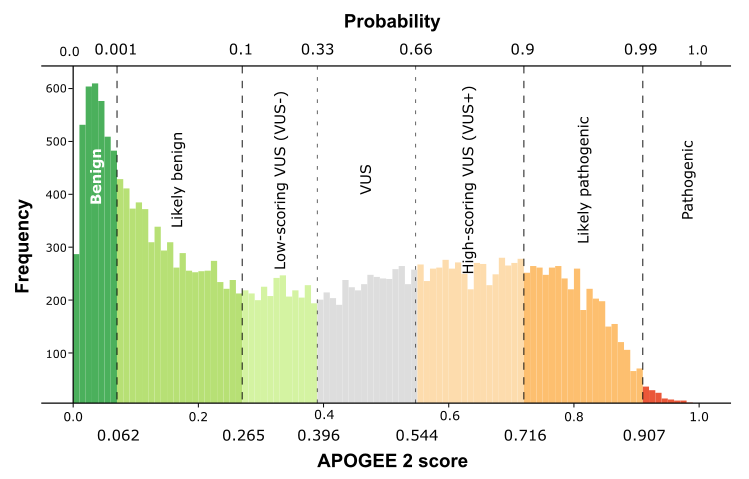
| APOGEE2 |
Pathogenic |
Likely-pathogenic |
| APOGEE2 score |
0.930438486242685 |
0.856900756978538 |
| CAROL |
deleterious |
deleterious |
| CAROL score |
1.0 |
1.0 |
| Condel |
neutral |
neutral |
| Condel score |
0.15 |
0.06 |
| COVEC WMV |
deleterious |
deleterious |
| COVEC WMV score |
2 |
2 |
| MtoolBox |
deleterious |
deleterious |
| MtoolBox DS |
0.86 |
0.87 |
| DEOGEN2 |
Damaging |
. |
| DEOGEN2 score |
0.844405 |
. |
| DEOGEN2 converted rankscore |
0.96400 |
. |
| Meta-SNP |
Disease |
. |
| Meta-SNP score |
0.907 |
. |
| PolyPhen2 transf |
low impact |
low impact |
| PolyPhen2 transf score |
-3.55 |
-3.55 |
| SIFT_transf |
medium impact |
medium impact |
| SIFT transf score |
-0.01 |
-0.27 |
| MutationAssessor transf |
medium impact |
high impact |
| MutationAssessor transf score |
1.93 |
2.21 |
| CHASM |
Neutral |
Neutral |
| CHASM pvalue |
0.64 |
0.68 |
| CHASM FDR |
0.8 |
0.85 |
| ClinVar id |
9691.0 |
. |
| ClinVar Allele id |
24730.0 |
. |
| ClinVar CLNDISDB |
MONDO:MONDO:0044970,MedGen:C0751651,Orphanet:68380|Human_Phenotype_Ontology:HP:0001086,Human_Phenotype_Ontology:HP:0001112,MONDO:MONDO:0010788,MedGen:C0917796,OMIM:535000,Orphanet:104 |
. |
| ClinVar CLNDN |
Mitochondrial_disease|Leber_optic_atrophy |
. |
| ClinVar CLNSIG |
Likely_pathogenic |
. |
| MITOMAP Disease Clinical info |
LHON |
. |
| MITOMAP Disease Status |
Cfrm [LP] |
. |
| MITOMAP Disease Hom/Het |
-/+ |
./. |
| MITOMAP General GenBank Freq |
0.0033% |
. |
| MITOMAP General GenBank Seqs |
2 |
. |
| MITOMAP General Curated refs |
22879922;11133798;33779865;16380918;15972314;17122117;20301353;21457906;29253894;21397051 |
. |
| MITOMAP Variant Class |
disease |
. |
| gnomAD 3.1 AN |
. |
. |
| gnomAD 3.1 AC Homo |
. |
. |
| gnomAD 3.1 AF Hom |
. |
. |
| gnomAD 3.1 AC Het |
. |
. |
| gnomAD 3.1 AF Het |
. |
. |
| gnomAD 3.1 filter |
. |
. |
| HelixMTdb AC Hom |
. |
. |
| HelixMTdb AF Hom |
. |
. |
| HelixMTdb AC Het |
. |
. |
| HelixMTdb AF Het |
. |
. |
| HelixMTdb mean ARF |
. |
. |
| HelixMTdb max ARF |
. |
. |
| ToMMo 54KJPN AC |
. |
. |
| ToMMo 54KJPN AF |
. |
. |
| ToMMo 54KJPN AN |
. |
. |
| COSMIC 90 |
. |
. |
| dbSNP 156 id |
rs199476106 |
. |